
单细胞分析进入多组学时代

多组学数据正越来越多地与空间信息相结合。
快速增长的软件工具集合正在帮助研究人员分析多个巨大的“组学”数据集。
一只小鼠从受精卵成长为新生幼崽大约需要20天。Ricard Argelaguet等人对小鼠胚胎第4.5天至第7.5天之间细胞内的发育情况很感兴趣。在这期间干细胞转变为三个胚层:外胚层,发育为神经系统;中胚层,发育为肌肉和骨骼;内胚层,发育为肠道和内脏器官。
虽然研究人员可以通过观察单个细胞中表达的基因来轻松区分这三个胚层,但该团队希望有一个更细致入微的画面。因此,在2019年,研究人员将基因表达数据与其他两个信息来源相结合。第一个是甲基化(methylation):一种改变基因表达方式的化学修饰。第二个是染色质的可及性(chromatin accessibility):染色质(真核细胞核中蛋白质和DNA的多节复合体)的修饰如何影响DNA的哪些部分可被用于转录成RNA。两者都是表观遗传学的因素,即影响基因表达方式的非遗传因素。
结合这三个数据来源,发现了一些意想不到的现象:在没有外部刺激的情况下,胚胎干细胞会变成外胚层。Argelaguet表示,这是这篇论文最重要的贡献。它表明在表观遗传水平上存在着一种细胞命运规范的层次结构。英国剑桥巴布拉汉姆研究所(Babraham Institute)的计算生物学家Argelaguet是这项研究的四位第一作者之一,该研究由巴布拉汉姆研究所研究员Wolf Reik以及附近Hinxton的EMBL欧洲生物信息学研究所(EMBL-European Bioinformatics Institute)的John Marioni和海德堡德国癌症研究中心(German Cancer Research Center)的Oliver Stegle监督。
他们的结果解释了几十年来的观察,即胚胎干细胞在培养中会优先分化为神经元。Argelaguet表示,这是一个仅仅使用单一类型的数据不可能做到的发现。
基因组学爆发
过去十年见证了单细胞基因组学的爆发。分析基因表达方式的单细胞RNA测序(RNA sequencing, RNA-seq)是最常见的技术。其他方法则详细说明了甲基化、遗传变异(genetic variation)、蛋白质丰度(protein abundance)和染色质可及性等过程。
现在,研究人员越来越多地将这些方法以及由此产生的数据层结合到“多组学”实验中。例如,Argelaguet在一项名为scNMT-seq的技术中结合了基因表达谱、甲基化和染色质可及性。另一项技术CITE-seq能同时对转录和蛋白质丰度进行分析。而G&T-seq同时捕获了基因组DNA和RNA。
无论缩写是什么,所有这些技术都旨在收集使用任何单一方法都可能无法检测到的复杂生物学见解。但这项任务在计算上具有挑战性,并且对由此产生的结果数据的分析更是如此。一套快速发展的软件工具可以提供帮助。
几乎所有的单细胞研究都包含可视化,有时称为t-SNE或UMAP图,可将单细胞表示为二维平面上的点。研究这些点如何聚集或聚类,可以帮助研究人员辨别生物结构。但可视化的创建并不容易。
首先,单细胞数据集已迅速变得巨大。早在2019年,Argelaguet使用荧光激活细胞分拣机捕捉微孔板中的单个细胞,这使他每周只能分析200-300个细胞。现在,他可以处理数以千计的细胞,这部分归功于位于加利福尼亚州普莱森顿的生物技术公司(biotechnology company)10x Genomics开发的微流控平台。而由西雅图华盛顿大学(University of Washington)的基因组科学家Cole Trapnell和Jay Shendure监督的2020年人类胎儿基因表达图谱包括400万个细胞。结果基本上是一个包含800亿个条目的表格:由2万个基因组成的400万行细胞。
然而,Trapnell表示该矩阵中的绝大多数条目都是零。这代表了一个关键的统计和计算挑战,因为科学家们正在努力区分真正的零:例如,实际上没有表达的基因和由于样本处理或敏感性问题导致的遗漏。一种选择是使用缺失值填充方法,即从数据集中的类似细胞中“借用”数据来填补空白。正如Stegle所说,你的邻居告诉你一些关于未知的事情。
难度增加
aguet表示,将各种模式结合起来只会使难度增加。每一种技术的所有弱点、所有误差和所有挑战,通过将它们结合到一个多模态的检测中,都会变得更加严峻。
Argelaguet等人花了三个月来收集他们的数据集,并用了两年对其进行分析。马萨诸塞州剑桥市哈佛大学(Harvard University)的表观遗传学家Jason Buenrostro表示,他的团队对描述一项被称为SHARE-seq方法的研究中的某些计算花费了数周才完成。
瑞士苏黎世大学(University of Zurich)研究单细胞肿瘤生物学的Bernd Bodenmiller认为,增加的细节所带来的回报是,它有助于研究人员去理解生物学。他们可以利用现有的数据集来做到这一点,如人类细胞图谱及其1350万个细胞谱。在6月发表的一篇预印本中,Shendure实验室的研究生Chengxiang Qiu等人结合了来自4个已发表的图谱的140万个细胞,以了解一种细胞类型如何在小鼠发育的10天内从另一种细胞中产生。由此产生的“哺乳动物胚胎发育轨迹”(trajectories of mammalian embryogenesis)揭示了500多个可能在细胞类型规范中发挥作用的转录因子。
软件工具
Marioni表示,信息可以通过三种主要方式进行整合,这取决于数据集有哪些共同的特征(或“锚点”,anchors)。他还发表了一篇关于该主题的评论。“水平整合”(Horizontal integration)用于相同类型的数据集,例如两个RNA-seq数据集。Marioni认为,在这种情况下,基因可以充当锚点,因为在每个细胞群中检测的都是同一组基因。
“垂直整合”(Vertical integration)涉及从同一细胞中收集的数据集,如RNA-seq和染色质可及性。而“对角线整合”(diagonal integration)涉及在不相关的细胞群中进行的分子测量。Marioni表示,问题是,要使用的共同特征是什么?垂直整合的一种方法是将染色质可及性位点与它们所调控的基因联系起来,然后从数据中计算出可能的基因表达谱。
所以Marioni认为,基本上把它变成了一个水平整合问题,基因再次成为锚点。
Trapnell表示,整合数据集就像对齐DNA序列。假设你能用一种方式看到的细胞群在另一种方式下也是可见的,而且对于大多数细胞或细胞群来说,会有一个一对一的映射关系。诀窍是将这两组数据对齐,以便可以确信看到的任何差异不是由于无法找到相似之处所引起的。而这也与启发大多数序列比对算法的精神相同。
为了实现这一目标,研究人员已经开发了几十种工具。其中许多被收录在GitHub的社区驱动的“awesome-multi-omics”和“awesome-single-cell”列表中。
Satija小组的计算生物学家Tim Stuart表示,例如纽约基因组中心(New York Genome Center)的Rahul Satija团队开发的Seurat软件,能有效地将两个数据集的UMAP可视化对齐,创建一个“共享的低维”空间,这使得能够在另一个数据集中找到一个临近数据集,反之亦然。其他流行的选择包括:Argelaguet的MOFA,他将其描述为主成分分析(principal-component analysis)的“一种多组学整合”;Harmony,来自马萨诸塞州波士顿哈佛医学院的Soumya Raychaudhuri团队;以及LIGER,由密歇根大学(University of Michigan)安阿伯分校的Joshua Welch团队开发。根据Welch的说法,就像在线零售商可以挖掘他们客户的购买历史来识别用户可能想要的产品一样,LIGER使用“综合非负矩阵分解”(non-negative matrix factorization)来识别相关细胞和细胞集群。
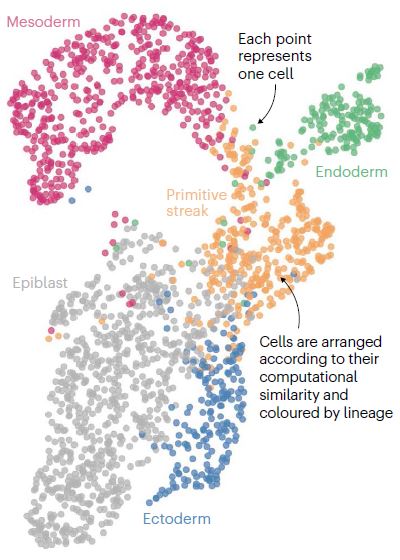
绘制UMAP
降维可视化使研究人员能够分辨出隐藏在细胞群中的生物结构。这个“统一流形近似与投影图”(uniform manifold approximation and projection, UMAP)代表了小鼠胚胎发育早期阶段研究中的1928个细胞。
空间黑客马拉松
面对这样一个快速增长的工具集,研究人员可能很难知道他们应该使用什么方法来解决哪些问题,以及如何去做。为了帮助弥补这些差距,马里兰州巴尔约翰霍普金斯大学(Johns Hopkins University)的Elana Fertig、波士顿哈佛大学陈氏公共卫生学院(Harvard T. H. Chan School of Public Health)的Aedin Culhane和澳大利亚墨尔本大学(University of Melbourne)的Kim-Anh Lê Cao组织了一次关于单细胞全能数据整合的虚拟会议。作为2020年6月举行的那次活动的一部分,组织者提供了三个精心策划的数据集,并要求与会者在一系列“黑客马拉松”(hackathons)中应用他们喜欢的任何算法和工作流程来集成和解释数据。与研究人员在几个小时或几天内密集地合作进行软件项目的面对面的黑客马拉松不同的是,这些是在一个月内举行的虚拟活动,合作者分散在全球各地。其中一次活动侧重于Argelaguet的小鼠胚胎数据集,其他活动则集中在空间数据集成问题。
Lê Cao表示,他们很想知道他们会在多组学中经历哪些挑战。他们认为最好将该领域的不同专家聚集在一起,看看专家们将如何进行单细胞多组学研究的分析。
传统的单细胞实验以牺牲位置信息为代价,详细记录了成千上万的分子。空间方法不需要分离步骤就可以捕捉到分子特征。通过将这两种数据类型分层,研究人员便可以计算出分离细胞的可能物理位置,或者用额外的分子细节来充实空间数据集。
Marioni认为,一个细胞如何决定它的命运,它将如何运作,是许多因素的组合。但非常重要的是细胞在胚胎中的物理位置:对它的机械压力、局部的信号环境、胚胎的形状,以及它如何在发育过程中发生变化。因此,如果我们想更好地了解细胞命运决定,在空间进行这些检测真的很有帮助。
在一项挑战中,研究人员得到了来自小鼠视觉皮层的空间和非空间的RNA表达数据集。然后,他们被要求使用在非空间数据中计算的细胞类型分配来识别空间数据中的细胞类型,其中每个细胞被识别的基因较少。第二个挑战是,是否有可能在非空间转录数据中确定细胞位置的基因表达特征。据Fertig所述,这个问题的答案是混合的。她表示,这取决于数据集和细胞类型。
加拿大汉密尔顿麦克马斯特大学(McMaster University)的统计学家Pratheepa Jeganathan解决了第三个挑战,其中涉及来自不同乳腺癌患者队列的蛋白质丰度数据。黑客马拉松参赛者的任务是整合部分重叠的蛋白质组学数据集、推断不存在空间数据的细胞的位置以及使用非空间数据来预测空间数据中未检测的蛋白质的表达水平。
Lê Cao表示,与会者主要通过重新利用现有算法来参加这些黑客马拉松。她的学生使用了一种名为偏最小二乘法(partial least squares)的机器学习方法,该方法最初是为大量基因组数据开发的。她表示她们正在积累知识和方法,并对它们进行调整,所以他们不是在做无用功。
Jeganathan使用文本主题分析(topic analysis),一种她在博士后期间改编的自然语言处理技术,来推断微生物群落在不同环境中的差异。在黑客马拉松中,她再次调整了该方法,以描述跨数据集的细胞的空间分布和组成。据Culhane描述,这种信息在临床上是有用的,因为肿瘤周围的免疫细胞的分布可以影响一个人对治疗的反应程度。Jeganathan认为,细胞的空间方向实际上对病人的生存是有参考价值的。
基因调控网络
两种组学数据类型对于确定细胞发育的分子机制特别有用。
单细胞RNA-seq数据确定了哪些基因在一个特定的细胞中表达,而染色质可及性检测则突出了调控区域。通过整合这些数据,研究人员可以识别作用于一个基因的调控元素、可能控制这些元素的转录因子以及这些转录因子的表达时间和空间。其结果是,研究人员可以通过探究这个基因调控网络来区分细胞的命运是如何被决定的。
Buenrostro和他的团队应用这一策略来展示染色质是如何在小鼠皮肤的细胞分化之前启动,或成为预备状态的。然后他们能够利用细胞的染色质潜力(chromatin potential)来预测单个细胞可能如何分化。据Buenrostro解释,染色质应该始终指向分化的方向。他的团队已经发布了一个名为FigR的软件包,以帮助定义这些网络。
密苏里州圣路易斯华盛顿大学(Washington University)的Samantha Morris团队的CellOracle,允许研究人员模拟阻碍或促进转录因子对细胞特性的影响。Morris与意大利米兰的研究人员合作,研究特定的转录因子如何影响人类胚胎中被称为中棘神经元(medium spiny neurons)的脑细胞的发育,而这是不可能通过基因操作来实现的。另外,她的团队通过计算修改了约200个转录因子,以确定哪些参与胚胎斑马鱼(Danio rerio)中轴中胚层的形成。轴向中胚层发育成脊索,脊索是支撑胚胎身体的骨骼杆。该软件预测,这些转录因子之一“noto”的缺失不仅会阻止已经明确的脊索发育,而且还会促进另一种发育结构的生长,而这是未知的。当他们在实验室中敲除noto时,这正是他们所看到的情况。Morris表示,他们能够预测这种敲除的新表型,然后用单细胞RNA-seq实验验证了这一点。
不遗余力
随着单细胞多组学领域的加速发展,新工具正以令人眼花缭乱的速度出现。如果细胞信息可以通过测序捕获,单细胞生物学家正在将其纳入他们的实验。
今年6月,美国和日本的研究人员描述了一种同时捕获三种信息的方法:染色质可及性、细胞表面蛋白丰度和细胞谱系,其中最后一种信息是利用线粒体DNA检测的。
该团队最初将其方法称为ASAP-seq。但在论文修订期间,10x Genomics公司发布了一个新的微流控套件,以简化从同一细胞中收集基因表达和染色质可及性数据的工作,研究人员决定将该套件与ASAP-seq结合起来,以折叠出另一层信息:转录。
该团队将这种方法命名为DOGMA-seq,这是对“分子生物学的中心法则”(central dogma of molecular biology)的致敬,即DNA被转录成RNA,而RNA被翻译成蛋白质。除其它事项外,该技术揭示了骨髓分化过程中的系谱偏差。
加利福尼亚州斯坦福大学的计算生物学家、该团队的成员Caleb Lareau表示,引入一种新的检测方法作为修订实验这一事实首先说明了单细胞领域正在以惊人的速度发展。
研究人员只能努力跟上。发展的速度如此之快,以至于Buenrostro开玩笑称,他的学生在看每篇新发表的文章时头脑都会“崩溃”,因为他们争先恐后地想知道这些文章对他们的研究有什么影响。
Lareau表示,他们团队已经抢先一步命名了他们的DOGMA-seq继任者。他们的工作名称是什么?“Kitchen-seq”,意思是:怎么可能不遗余力地测序任何东西?
原文检索:
Jeffrey M. Perkel. (2021) single-cell analysis enters the multiomics age. Nature, 595: 614-616.
郭庭玥/编译



{kind=link}