
关注冷冻电子显微镜的准确性
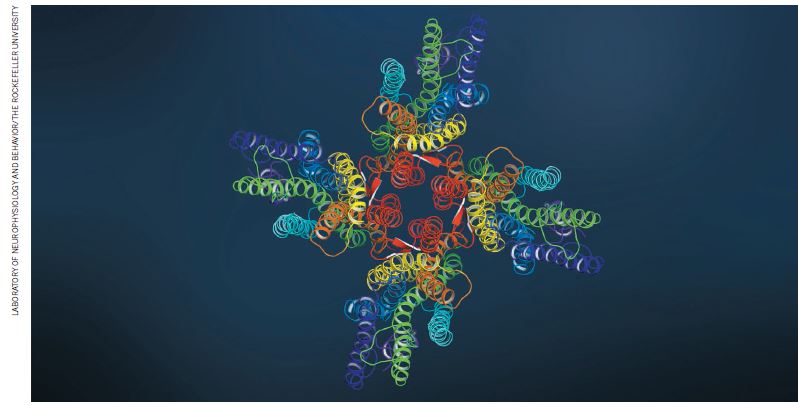
X射线晶体衍射成像难以对图中昆虫气味受体等离子通道的结构进行成像。冷冻电子显微镜则能得到清晰的结构图片。
随着蛋白结构成像技术的分辨率越来越高,研究人员正在竞相开发工具来评估它们的准确程度。
冷冻电子显微镜(cryo-electron microscopy, cryo-EM)一度由于图像过于模糊,而被嘲笑为“只能看见一坨东西的技术(blobology)”。但现在,冷冻电镜分辨率大幅提升,可以对病毒颗粒和酶等多种结构进行高分辨率成像。2002年,上传到电子显微镜数据库(Electron Microscopy Data Bank, EMDB)的冷冻电镜图片数量仅为8,到2017年,这个数字增长到1106——且2017年该技术斩获了诺贝尔化学奖。
现在,冷冻电镜图像的质量已经可以与X射线晶体成像技术相媲美。X射线晶体衍射成像长期以来一直是解析蛋白质结构的主流技术。虽然一些特殊的蛋白及其互作无法用X射线晶体学进行成像,但却可以用冷冻电镜成像:例如,温度敏感的离子通道如何工作,表征神经退行性疾病中的病理蛋白,以及详细阐述病毒如何与抗体相互作用等。因此,许多经验丰富的晶体学家开始放弃X射线晶体衍射法,转而投向冷冻电镜成像技术的怀抱。
冷冻电镜解析分子结构的论文也越来越多(参见“打破瓶颈”)。但是,一些研究人员担心,不是每个人都知道如何评估模型的准确性。部分研究者正在呼吁采用新的流程和工具来提高冷冻电镜成像的准确性。
打破瓶颈
更好的验证工具和实践并不是冷冻电镜普及的唯一阻碍;许多研究人员无法生成足够清晰的图像,甚至无法开始成像。
纽约结构生物学中心(New York Structural Biology Center)电子显微镜联合主任Bridget Carragher指出,真正的瓶颈是标本制备。幸运的是,她共同开发的一项技术可能会打破这一瓶颈。
Bridget Carragher和该中心的另一位联合主任Clint Potter开发了具有液滴感应摄像头和压电设备的机器人,这些设备与喷墨打印机中的相同零件相似。一个机器人将小片样本放入一个网格中,另一个机器人将样品快速均匀地浸没到冷冻剂中,从而形成薄而均匀、适合成像的冷冻片。网格本身被纳米线覆盖,这些纳米线以受控的方式吸走多余的冷冻剂。这降低了蛋白质在空气-水界面处卡住的可能性,一旦卡住,蛋白质会发生变性,或倾向于采取某一特定构象,而非各角度成像所需的随机构象。
该系统名为Spotiton,允许用户更快地准备更多样品,并使用更少的蛋白质样本,并确保一份样本可以用更多次(方便对同一样本进行多次成像)。Carragher和Potter已将Spotiton授权给英国的TTP Labtech公司,该公司计划在明年左右以Chameleon的名义将该系统商业化。
创建标准
英国剑桥分子生物学医学研究委员会实验室(Medical Research Council Laboratory of Molecular Biology)成员、因冷冻电镜获得诺奖的Richard Henderson表示,冷冻电镜的技术进步很大程度上得益于更好的电子探测器和图像处理技术。不过,该领域仍然缺乏那种标准化的工具来构建可靠的结构模型。这导致了很多问题,现在需要的是更好的标准,鼓励研究人员将更多的精力投入到他们的模型构建中。
德国哥廷根马克斯普朗克生物物理化学研究所(Max Planck Institute for Biophysical Chemistry)的电子显微镜专家Holger Stark表示,现在冷冻电镜领域的竞赛重点在于得到分辨率更高的结构,这并不利于提高数据的可靠性。一些已发表的结构描述了原子水平的精确度,但没有承认结构的某些区域纯属“幻想”,缺乏数据来支持任何特定的解释。很多信息只是研究者放入原子坐标里的噪音。
加州克里普斯研究所(Scripps Research Institute)的结构生物学家Gabriel Lander指出,毫无疑问,冷冻电镜能够实现梦幻般的发现,并且许多结构都是可靠的。但他警告说,许多研究人员过于迅速地认为结构中的所有细节都是正确的。因此,他解释说,有人根据得到的蛋白结构设计一种突变形式来研究蛋白功能,或在预料之外的位点看到了配体结合,都会导致数月的实验失败。Lander担心,过度解读可能会坏了冷冻电镜的名声。
分辨率问题
蛋白质结构通常由单一因素判断:分辨率,即结构显示的细节水平。该指标在结晶学中很容易确定,但在冷冻电镜中则不然。
在晶体学中,紧密堆积的分子的高度有序晶格通过X射线束旋转,并且所得图像的分辨率可以直接由偏转光子产生的衍射图案计算。然后研究人员将这些衍射模式转换为电子密度的“图谱”,将其与蛋白质序列结合起来构建模型。该模型能表征蛋白质的特定化学结构单元如何折叠成片状和螺旋状(分辨率约5埃),以及氨基酸的侧链如何定位(分辨率在3.5埃左右)。大的、松散的分子往往不会形成有序的晶体,因此体积越小、结构越紧凑的蛋白质,就越适合X射线晶体衍射成像。
在冷冻电镜中,蛋白质和其它大分子复合物在薄薄的水层中快速冷冻,理想情况下冷冻片不会比蛋白质本身厚。用低能电子照射该冷冻片会在探测器上产生单个粒子的2D图像——由散射电子投射的模糊阴影(图“冰中建模”)。然后对数千,甚至数十万充满噪声的图像进行计算分类,最后重建得到3D图谱。最后,使用其它类型的软件将蛋白质序列拟合到图谱中以创建模型。分子越小,图像越嘈杂,因此冷冻电镜往往最适用于较大的分子。

为了避免把噪音误认为信号,研究人员通常将粒子分成两个子集,并从每个子集构建 “半图”。这两张地图之间的相关性可用于计算分辨率。但弗吉尼亚大学(University of Virginia)的结构生物学家Edward Egelman指出,这是一个不完美的标准。这种方法衡量的不是分辨率本身,而是一致性。由此必须审慎看待得到的结果。事实上,Egelman表示,盲目追求高分辨率有时会导致研究人员犯低级错误——例如将分辨率报告为百分之一,甚至千分之一的埃,事实上这种分辨率在冷冻电镜上毫无意义。
而且,并非所有错误信号都是随机噪声。Egelman已经证明系统伪像(例如将不存在的圆柱体计算添加到两个半图中)可以极大地(并且错误地)改善一个结构的表观分辨率。
有时,研究人员实际上利用得到的结构模型反向计算电子密度图,然后从中挑选最能支持模型的粒子数据。德国哥廷根大学(University of Göttingen)的晶体学家Piotr Neumann表示,这是一种人为引入的偏差。虽然这种作弊是不可接受的,但可以理解。另一种更常见的方法是创建蛋白质预期整体形状的“面具”,并用它来排除部分图像。合理使用“面具”,能提高信噪比;过度使用,则会引入偏差。
调整以适应
结构生物学家开玩笑表示,分子结构解析的相关文章中使用的分辨率多为2.9埃,而不是3.0埃——这是过度分析的明显症状。Lander则指出,即使没有过度分析,使用单一一个数字来描述蛋白质也存在问题。因为单一数字掩盖了这样一个事实:冷冻电镜图谱的质量差异很大,而且通常质量最差的往往是最灵活、生物学领域最感兴趣的蛋白区域。Neumann指出,没有哪一个衡量标准是足够完美的,所有指标都可能存在偏差或不完全可靠。所以,我们需要同时使用多个指标。
今年早些时候,Neumann等人开始记录蛋白质数据库中蛋白质结构模型与EMDB中相应电子图谱的匹配程度。他们发现,对于所检查的565个结构中,超过四分之三的结构的匹配程度仅为低或中,这表明大批蛋白结构模型需要以怀疑的眼光看待。
至少一些药物开发商对这些模型持审慎态度。瑞士巴塞尔诺华生物医学研究所(Novartis Institutes for Biomedical Research)冷冻电镜团队负责人Christian Wiesmann表示,在查看与小分子结合的蛋白质模型时,他通常从EMDB下载图谱,以评估其他研究人员如何设计小分子与这些蛋白质发生作用,然后基于自己的经验作出判断。Wiesmann指出,他有好几次发现模型与实际结构的差异——这些差异可能影响药物设计。
并非每个研究人员的研究都需要深入到这种复杂程度的结构内部。但即使他们需要,他们也很难找到图谱。马里兰州美国国家癌症研究所(US National Cancer Institute)的结构生物学家Alex Wlodawar指出,在发表论文时,作者必须将论文中涉及的图谱存放在EMDB中,但是这些图谱的网址通常没有获得充分的注释。Wkidawar在高分辨率下比较了晶体和冷冻电镜得到的分子结构,他发现后者往往过于“乐观”。研究人员可能会在没有说明锐化参数的情况下提供原始图片,或不告知使用的、用于排除部分数据的“面具”。此外,很少有人提交验证分析时使用的“半图”。
展望
管理EMDB的Ardan Patwardhan表示,与模型一样,不同的图谱质量差异很大。目前已有一些自动化和半自动化的工具套件,可以帮助研究人员将2D冷冻电镜图像转换为3D图谱。为了帮助评估这些工作流程,EMDB已经开展了多次验证竞赛。它发现最大的可变性并非来自软件包,而是来自用户的经验水平。经验较少的组使用默认参数;最好的团队为数据量身定制参数。表现好的团队可以得到清晰的侧链图像,而表现差的团队得到的二级结构都是模糊的,哪怕两组使用的原始图片一模一样。
现在,研究人员正在呼吁使用更好的方法来验证冷冻电镜图谱和模型——原始图像数据可能有用。 2014年,Patwardhan和他在英国剑桥欧洲生物信息学研究所(European Bioinformatics Institute, EBI)的同事创建了电子显微镜公共图像档案(Electron Microscopy Public Image Archive)。该库是目前175个原始图像数据存储库中最大的一个,总容量超过12太字节,全部下载大约需要5天。
更好的、表示不确定性的方法也能帮助提高准确性。Lander提出,研究人员提供了一系列模型,以更好地说明可能跟数据匹配的多种结构。伦敦大学(University of London)的计算结构生物学家Maya Topf创建了名为TEMPy的软件,该软件以氨基酸而非整个结构的尺度来衡量模型的质量。Topf表示,虽然目前评估还不是强制性的,但研究界开始期待这种评估。这种意识在增强。越来越多的研究者在论文中指出了局部分辨率。
尽管如此,冷冻电镜还有很长的路要走,才能达到X射线晶体学的成熟程度。EBI的结构生物学家Gerard Kleywegt指出,人们需要深刻意识到,必须验证数据和模型,特别是因为该领域吸引了许多缺乏经验的新从业者。当然,冷冻电镜和X射线晶体学在一些方面有本质上的区别:晶体学以刚性构象捕获蛋白质,而冷冻电镜可以显示更自然、自然模糊的构象。冷冻电镜技术的改进将需要更好的方法、更广泛的共识和更好的实践——所有这些都需要时间。Kleywegt指出,一个验证工作组早于2010年9月就召开了会议,以制定建议。自那以后,冷冻电镜领域发展迅速,后续会议也在开展中。目前2019年会议的筹划工作已在进行中。
原文检索:
Monya Baker. (2018) Cryo-electron microscopy shapes up. Nature, 561: 565-567.
张洁/编译



{kind=link}