
Starfish助推单细胞RNA测序
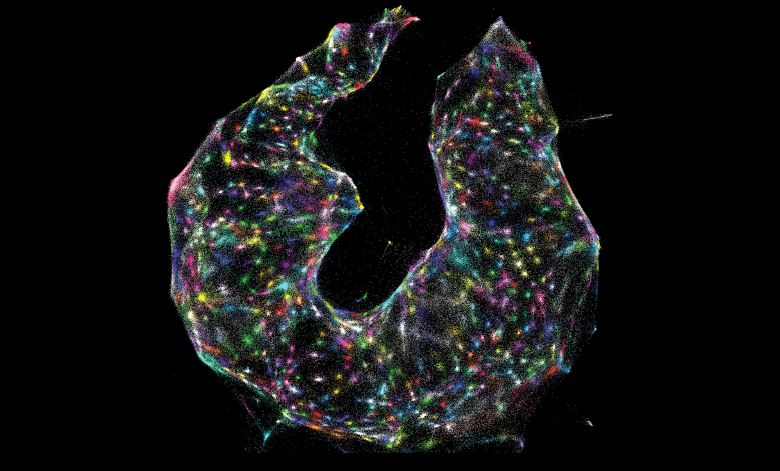
每一个点都是一个单独的RNA分子,由其与其它RNA的接近程度来定位。这种成像方法被称为DNA显微镜,因为它使用了DNA测序。
将数据分析工具Starfish与精确定位RNA细胞位置的技术相结合,可以增加原位转录组的空间细节。
对于电影爱好者来说,《太空堵塞》(Space Jam)是一部1996年的喜剧电影,讲述卡通人物兔八哥和篮球运动员Michael Jordan与动画外星人对决的故事。对于华盛顿西雅图艾伦脑科学研究所(Allen Institute for Brain Science)的神经科学家Ed Lein来说,这是一个以生物信息学为主题的会议的名字——意味着一次“黑客头脑风暴”。
4月,大约40名计算和转录生物学家齐聚艾伦脑科学研究所。他们来这里是为了喝咖啡,写代码,以及一个共同的目标:找出原位(或空间)转录组学领域各种技术的优点、缺点和分析挑战。
原位转录组学是一系列技术的组合,包括多重抗误差矫正荧光原位杂交技术(multiplexed error-robust fluorescence in situ hybridization, MERFISH)、序贯荧光原位杂交(sequential fluorescence in situ hybridization,seqFISH)、STARmap(Spatially-resolved Transcript Amplicon Readout Mapping,一种可以在3D组织中实现单细胞RNA测序的技术)和荧光原位RNA测序(Fluorescent in situ sequencing, FISSEQ)等多种技术在组织环境中绘制细胞的基因表达模式。有些人依靠杂交——短核酸探针在拥挤的细胞环境中寻找互补物的能力——而另一些人则基于DNA测序。但所有这些都产生了概念上相似的数据——基因表达值与细胞的x和y坐标相匹配。
这些数据可以揭示可能被忽视的细胞间关系,例如哪些细胞在与哪些细胞交谈,以及它们的位置与结构特征之间的关联,以及它们与目标细胞之间的相对位置。正如计算和系统生物学家、麻省理工学院-哈佛大学博德研究所(Broad Institute of MIT and Harvard)人类细胞图集(Human Cell Atlas, HCA)项目的创始联席主席Aviv Regev所指出,根据周围的分子信息,他们可以判定目标RNA分子的身份。
但该领域的发展如此之快,研究人员可能难以决定使用哪种方法。大量的数据分析算法、管道和文件格式会使数据分析和比较变得困难。Lein表示,该领域的现状是技术开发过于旺盛。
在慈善组织陈-扎克伯格计划(Chan Zuckerberg Initiative, CZI)的资助下,以及HCA的赞助下,Lein等人于2017年成立了一个研究联盟SpaceTx——spatial transcriptomics(空间转录组学)的缩写,对不同的方法进行基准测试。与此同时,CZI的程序员开始建立一个统一的数据分析工具和文件格式,名为海星(Starfish),以促进HCA的努力,并帮助更广泛的转录生物学界。(加州红杉市领导CZI计算生物学研究的Jeremy Freeman解释说,Starfish这个名字“有点像个笑话”。RNA单细胞测序的许多空间方法依赖于FISH(荧光原位杂交)。在编程中,星号表示通配符。取这个名字就像开玩笑,表示RNA测序都依赖于FISH技术。)
Starfish是一个开放源代码的软件套件,可以读取图像文件、确认和消除图片中的噪音,在9种不同的实验策略(还有两种正在开发中)中找到代表RNA分子的斑点并识别它们。Lein指出,这次头脑风暴的目的是让开发人员和用户——空间转录组专家——聚在一起讨论,排除故障,并改进他们的方法。在此过程中,研究小组发现了细微的差异,这些差异可能会使那些想在不同实验中比较数据的人感到困惑。该小组还为如何掌握不断增加的技术集提供了一个模型。
原位转录组学
研究基因表达的研究人员通常是在整体水平上进行的,从一块组织中提取RNA,然后对其进行整体分析。在过去的十年里,像Drop-Seq这样的单细胞方法使得研究人员能够以牺牲空间细节为代价来探索细胞之间的差异。
这就是原位转录组学的起源。这些技术主要使用荧光显微镜和DNA测序来揭示组织内部细胞的类型和丰富的RNA分子。由此,研究人员可以计算出存在的细胞类型、它们的空间排列以及它们之间的关系。
Regev表示,这就像一系列水果甜点,如果所有的大块基因组学都是混合果汁,那么单细胞基因组学就是水果沙拉,而空间基因组学就是水果馅饼。如果你从上面看一个水果馅饼,所有的水果都是按照特定的模式排列的。
根据这种方法,这些数据就像夜空中的星星,或者五颜六色的艺术品。例如,由卡罗林斯卡研究所(Karolinska Institute)的Sten Linnarsson实验室的生物图像信息学家Simone Codeluppi领导的一项研究,使用了一种单分子FISH的循环变体,名为osmfish(由“awesome FISH”的发音而来),来绘制小鼠躯体感觉皮层的结构。最后的结果是根据基因表达模式对细胞进行着色,这张图片让人想起了彩色玻璃窗。
但这些数据也可以揭示出一些见解。在英国剑桥大学(University of Cambridge),神经生物学家和医生David Rowitch使用一种名为RNAScope的方法来研究小鼠大脑中星形胶质细胞的空间多样性和组织。Rowitch发现,星形胶质细胞“在皮层中采用类似于神经元的层模式,但与神经元的排布模式不一样。在加州理工学院(California Institute of Technology)研究单细胞生物学的Long Cai和他的团队使用一种叫做seqFISH+的策略来识别编码相邻细胞表面相互作用蛋白质的转录物。
增加清晰度
seqFISH+和RNAscope都依赖于核酸杂交,利用荧光标记的分子照亮细胞中的目标序列。其它方法则使用DNA测序,甚至质谱。
目前已经有十多种空间转录组学方法,其中6种是2019年开发的。它们在可检测的RNA数量、空间分辨率和可探测的细胞数量上存在差异,但都提供了单细胞转录组无法提供的空间定位细节。Regev指出,但是空间方法也有缺点。例如,显微镜检查速度慢(有时需要数周的连续成像)、成本高和技术要求高。许多方法只能访问细胞转录组的一个预先定义的部分,并且会限制可检测的细胞数量。
在试图选择正确的方法时,研究人员可能会不知所措。SpaceTx旨在提供一些清晰的信息。
CZI的一位发言人指出,该项目是CZI过去两年半在HCA和辅助项目上花费约1亿美元的一部分。每个小组——总共有19个——将自己的方法应用于艾伦研究所准备的人类和小鼠大脑的相同样本。现在,Lein等人,以及更广泛的计算生物学界,正在对比结果,看看这些方法是如何比较的,以及哪种方法最适合特定的环境。
Lein表示,这实际上是非常不寻常的。通常,研究人员致力于开发出最好的方法,发表并继续研究。但是对于SpaceTx,他们试图把所有人聚集在一起,并指出这些方法都是有用的,但是他们需要了解每个方法的用途,以及它们各自的相对优势。
但是这样做会带来一个计算问题,因为不同的方法产生不同的数据类型。例如,一些基于杂交的方法为每个转录本分配不同的颜色,而其它方法则使用多种颜色作为条形码。一些实验室通过跟踪每幅图像中的荧光点,然后监测它们在不同次成像之间的强度来识别RNA,而另一些实验室则通过测量每个像素的强度,将这些强度值与可能的条形码列表相关联,以确定是否存在RNA。这些图像在磁盘上的组织方式以及用于注释它们的“元数据”也可能有所不同。
英国辛克斯顿欧洲生物信息学研究所(European Bioinformatics Institute)的生物信息学家Matthew Green表示,这种不相容性会阻碍研究。即使他们不这样做,研究人员也常常很难安装同事的分析软件(由于这种软件需要复杂的计算需求和依赖性)。Green指出,空间研究所产生的海量数据可能令人生畏,Linnarsson的自动化osmfish设备每天产生2兆字节的图像,而他们组的SpaceTx相关工作已经产生了大约25兆字节的图像。
太空堵塞
CZI的Deep Ganguli和Ambrose Carr领导的一个计算生物学家和软件工程师团队开始创建一个用于原位转录组分析的标准文件格式和管道。该团队甚至参观实验室,和生物信息学家交谈,以了解他们的工作流程。Freeman指出,至少有一个实验室的研究生告诉Deep,这太棒了,因为以前都没人会看他的代码。
4月的头脑风暴为Starfish团队提供了一个让生物学家们试用这个软件的机会,同时也为研究人员和编码人员提供了一个交流机会,让他们面对面学习,而不是通过代码共享平台Github上的bug报告来交流。
负责CZI Starfish开发的Justin Kiggins表示,他们能够帮助所有这些人,让他们在Starfish上处理数据,以帮助他们的科学研究。这让Kiggins的团队对差距和挑战有了至关重要的了解。
加州大学(University of California)生物工程研究生Matt Cai开发了一种名为DARTFISH的方法。Cai表示,他在4月的“黑客头脑风暴”上有两个目标:与其它空间转录组交流想法,并与Starfish保持同步。Cai等人有自己的内部分析方法,但这些方法的编写方式不便于人们使用。不过Starfish不一样,它是为整个科学界而开发的。
对于Green来说,这次会议和他参加过的任何其他会议都不一样。尽管多年来他参加过多次会议,他表示,他从未参加过这样的会议,每一次谈话都像是一次大规模的信息交流。非常刺激。
Lein表示,每一个小组都成功地将一个样本数据集转换成了Starfish格式,数据生成也在进行中。但软件本身仍然在继续开发中。英国维康信托桑格研究所(Wellcome Sanger Institute )的程序员Aleksandra Tarkowska指出,由于“数据的复杂性”,她无法将数据集转换为Starfish格式,并将不同的视野排列成统一的图像。Long Cai实验室的软件工程师Nico Pierson报告了软件的“斑点解码器”(spot decoder)的问题,该算法将荧光模式与条形码相匹配,但它无法处理seqFISH+数据的密度。Pierson指出,对于他们的数据,转化效率非常低,可能只有10%。
尽管如此,与会者还是称赞这次活动提供了程序员和生物学家交谈的机会。程序员带来了一堆错误报告和特性请求,其中一些可以当场解决。研究人员带着新的更好的数据分析方法回到实验室。例如,Codeluppi发现了一种“分割”策略,用于计算识别图像数据中的细胞边界,特别是对于小体积细胞。
Matt Cai表示,他的实验室现在经常在自己的计算管道旁边运行Starfish来比较性能。但其他人可能不愿意放弃他们精心设计的内部管道。因此,Starfish可以在尝试实施其它研究方法的实验室中找到最大的应用。
哥伦比亚大学(Columbia University)扎克曼大脑行为研究所(Zuckerman Mind Brain Behavior Institute)的分子神经学家Abbas Rizvi指出,有了这么多不同的方法,以计算的形式把它们联系在一起真的很有价值,他认为Starfish会有助于实现各种方法的联系。Rizvi是HCA项目的成员,该项目利用部分空间方法构建了人类脊髓图谱。
Rizvi还指出,这让他想起了单细胞转录组学的早期阶段,要让实验成功已经足够困难了,但是分析数据并从数据中挖掘有价值的信息的方法也让人兴奋。Starfish就在做这件事。
更多阅读:
RNA研究方法
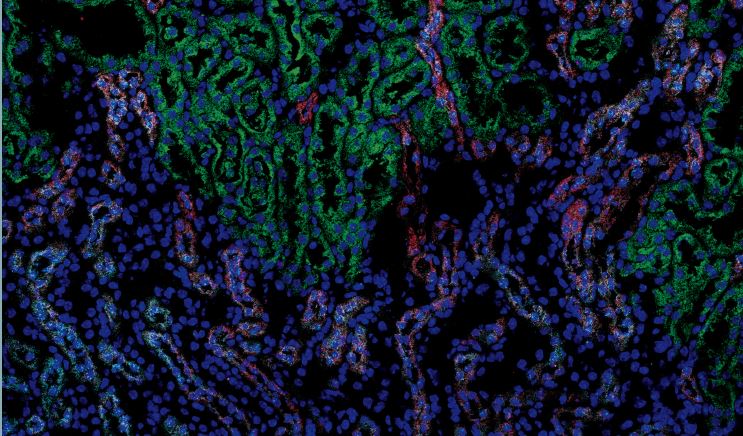
用荧光探针捕捉到的小鼠肾脏的图像。
目前有十几种原位转录组学方法,包括:
APEX-Seq:除非一个细胞被染色,否则在显微镜下它看起来毫无特征。因此很难确定特定RNA的位置。APEX-Seq将APEX2酶定位到一个特定的细胞“地址”,并用它标记附近的RNA。通过分离和测序这些RNA,研究人员可以分析单个细胞域的转录物。
DNA显微镜:DNA显微镜融合了分子和计算技术,根据每个分子的邻居推断出每个分子的位置,就像利用无线电台发射器的覆盖范围绘制美国地图一样。博德研究所Aviv Regev实验室的博士后研究员Joshua Weinstein指出,他们正在采集生物分子样本,并将每一个RNA转化为一个无线电发射塔。完整组织中的RNA在适当的位置被放大,创造出更大的核酸“扩散云”。当云与邻近的云接触时,就产生了一个独特的特征,研究人员可以利用DNA测序“读取”来重建样本的分子结构。
IMC:成像大细胞术(Imaging mass cytometry),通常用于定位细胞中的蛋白质,也可以用来精确定位少数的RNA。该方法将一种称为RNAScope的技术与质谱联用,以揭示免疫细胞释放的生长因子信号。
INSTA-Seq:原位可及性测序(In situ transcriptome accessibility sequencing, INSTA-Seq)是荧光原位测序的一个变型。该方法使用连接法进行序列测定,在原位识别RNA分子的短条形码,然后利用专有的化学照明方法提取这些RNA,并再次进行序列测定,以读取它们的全长。该方法的开发者,纽约冷泉港实验室(Cold Spring Harbor Laboratory)的分子遗传学家Je H.Lee表示,由于产生这些较长的读数所需的合成步骤可能会被蛋白质或其它RNA分子的结合等因素所阻断,因此该方法可以提供对“空间外显子”的深入了解。
RNAscope:在加州纽瓦克市,RNAScope被Advanced Cell Diagnostics公司商业化。RNAscope是一种基于原位杂交的方法,利用信号放大来提高每个目标RNA的亮度。12种RNA可以通过3轮成像来区分。
SeqFISH+:SeqFISH+结合了荧光条形码、“假染色”方法和多轮杂交,以“稀释”细胞RNA,使它们更容易被分析。在每个细胞中可以检测到多达10,000个不同的RNA。
Slide-seq & HDST:组织样本被排布在空间分辨率级别/二维码编码的磁珠上。每个细胞的RNA与细胞的“邮政编码”相关联。Slide-SEQ使用10微米大小的珠子(小到足以解决小鼠大脑一个细胞厚的特征)。高密度空间转录组使用2μm珠进行亚细胞分离。
STARmap:STARmap是组织透明技术、RNA扩增和DNA测序的结合,可以在其它不透明组织中识别多达1,020种RNA。每一个RNA被分配一个五碱基的基因识别条形码,通过序列连接读出。
原文检索:
Jeffrey M. Perkle. (2019) Starfish enterprise: finding RNA patterns in single cells. Nature, 572: 549-551.
张洁/编译



{kind=link}