
单细胞组学
前言
细胞作为生命最为基本的一个单元概念,是生命活动的基石。尽管生物学家在显微镜下工作了将近180年,但我们对细胞仍然不甚了解。本文着重探讨了研究人员如何通过观察单独的细胞来了解细胞的性质——存在多少种不同的细胞、它们做了什么以及它们如何发生改变。
我们期望有有效的技术手段,可以完整地检查单个细胞的成分,包括在细胞,甚至分子水平上鉴定和治疗疾病。对病理学理解的推进将使我们能够预测基因是如何使个体易患疾病,并帮助预防和治疗疾病的。这对于诸如癌症等疾病尤其重要,其通常具有极其多变的遗传组合物,从而在单一肿瘤内导致不同的基因表达谱。尽管缩小自身的技术仍然是虚构的,但我们在单细胞水平上想象基因如何行动的能力却不是,我们期待着扩大对人体的认识。
本文详细描述了谱系追踪的成果,揭示了复杂的生物体是如何由一个细胞,然后是两个,然后是四个,看起来相同的胚胎细胞构建而成的。单细胞组学研究比常规的细胞群体研究可以揭示更多细胞类型和亚群的多样性。这就是为什么马萨诸塞州Broad Institute的Aviv Regev等科学家建立人类细胞图谱计划,并积极参与对人体内各种细胞的分类的原因。
这样的工作有着重要的治疗意义,因为跟踪不同肿瘤细胞之间的差异可以指导治疗。此外,最近的技术进步使我们能够在单细胞水平上鉴定和可视化RNA转录物、蛋白质和其它细胞成分。这揭示了免疫系统、大脑和胚胎发育过程,并且将彻底改变我们对整个人体的认知。
近年来,已有越来越多的科学家进入单细胞分析领域,其中包括经典的细胞生物学、发育生物学、基因组学和计算生物学领域。而随着研究单细胞的技术的不断增加,人们将需要复杂的分析工具来分析和理解结果。正如细胞理论为生物学上的非凡进展提供了支持依据,单细胞分析显然将为科学家开拓新的前景。

个体细胞有遗传“指纹”的能力,使我们能够发现它们的微妙差异。
图片来源:https://d2ufo47lrtsv5s.cloudfront.net/content/sci/358/6359/56/F1.medium.gif
人类细胞图谱计划
细胞是生命的基本单位,Aviv Regev教授长期以来一直在寻找探索复杂的基因网络在单个细胞中的运作机制的方法,并想了解各个细胞中这些网络有何差异,以及最终各种细胞群体如何协同工作。这些问题的答案将从本质上揭示细胞如何构建复杂的生物体,如人类。在麻省理工学院和哈佛大学麻省理工学院工作的Regev和Levin对来自小鼠骨髓的18个看起来相同的免疫细胞的RNA进行了测序,结果发现其中一些细胞与其余细胞的基因表达模式截然不同。这些细胞感觉就像两个不同的细胞亚型。
这使Regev想要进一步开展深入的研究,即使用单细胞测序来了解人体内存在多少种不同的细胞类型、它们在哪个部位,以及它们如何发挥作用。Regev的实验室同时对18个细胞进行检测,一共测定了数十万个RNA的序列,并将单细胞分析与基因组编辑技术结合起来,以了解关键调控基因被抑制时会发生什么情况。
2016年末,Regev帮助推出了“国际人体细胞图谱计划(Human Cell Atlas)”,该计划雄心勃勃,准备对人体中所有(估计37万亿个)细胞进行分类和测序(图“建立人类细胞图谱”)。
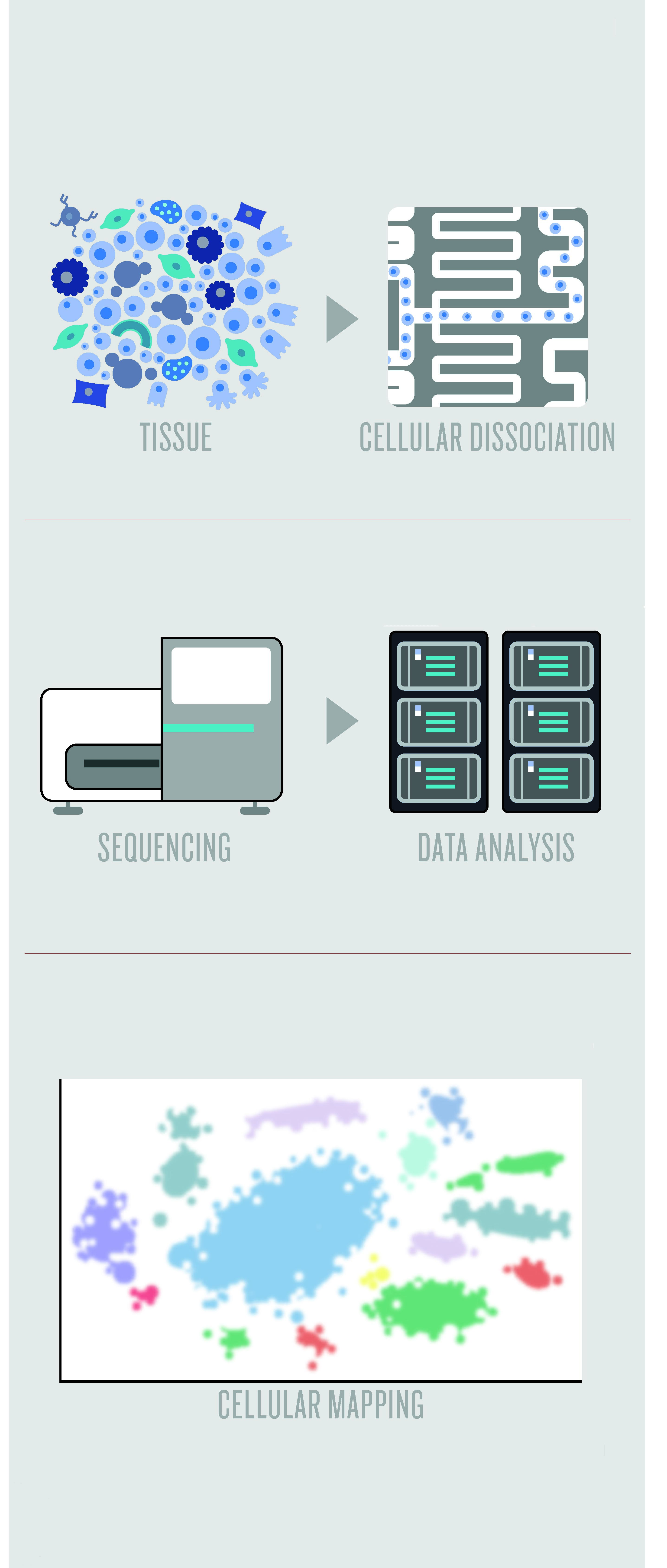
建立细胞图谱的方法。组织分离成细胞,进行测序和数据分析,最后完成投射。
图片来源:Anna Nowogrodzki. (2017) The cell seeker. Nature, 547(1038): 24-26.
人体细胞图谱项目计划对人体各种细胞的RNA进行测序,然后使用这些基因表达谱将细胞分类,定义新的细胞,并绘制所有细胞及其分子在空间上的组织方式。该项目还旨在发现和表征人体内所有可能的细胞状态——成熟和不成熟、满负荷和充分工作状态——这需要更多的测序。科学家认为,人体内大约有300种主要的细胞类型,但是Regev怀疑有更多的状态和亚型需要探索。她还认为,单独的视网膜似乎就含有100多种亚型的神经元。在共同部署人体细胞图谱项目时,Regev已经召集了来自五大洲的28人的委员会,并协助组织了有500多名科学家参加的会议。
在最基本的层面上,人类细胞图谱必须包含一个全面的人类细胞参考目录,因为它们包含细胞稳定性和瞬时特征,以及它们的位置和丰度。此外,图谱必须提供一个坐标系统,从而在多个层面表达和协调概念,并且我们可以在这个图谱可的任何级别的放大视图中查看特征,并将高维信息折叠成更简单的视图。由于一个细胞可能包含2万个基因表达情况,所以我们需要能够降低维度的分析工具。此外,图谱应具有附加的坐标或注释,以表示组织学和解剖学信息(例如,细胞的位置、形态或组织),时间信息(例如,自暴露后个体的年龄或时间)和疾病状态。这些信息对于根据分子谱和关于细胞生物学、组织学和功能的背景来协调结果至关重要。
人类基因组计划对生物医学产生了重大影响,提供了一个全面的参考资料。我们可以很轻易地通过一个DNA序列查找答案,并从中挖掘独特的“特征”。人类细胞图谱可以为基础研究提供类似的临床相关应用的好处。这种细胞特性的概括将被广泛用于临床检测。例如,今天的全血细胞计数(CBC),一个有限数量的血液成分的普查,可以补充一个“CBC 2.0”,以提供有核细胞的高分辨率图片,包括每个的数量和活动状态与健康参考样本进行比较。类似的措施也应该可以用于其它组织。例如,可以分析来自溃疡性结肠炎或结肠癌患者的肠道活检组织,了解其包含的各种上皮细胞、免疫细胞、基质细胞和神经细胞的类型、反应、状态和位置。
单细胞组学技术
目前,单细胞生物学是一个热门话题。该领域的最前沿研究是单细胞RNA测序(scRNA-seq)。RNA测序的常规“批量”方法(RNA-seq)可一次处理数十万个细胞,并平均差异。事实上,没有两个细胞是完全相同的,scRNA-seq则可以揭示使每个细胞独特的微妙变化。它甚至可以揭示全新的细胞类型。Regev等人利用scRNA-seq探测2400多种免疫系统细胞后,发现了一些具有强烈T细胞刺激活性的树突状细胞。一种刺激这些细胞的疫苗可能有助于激活免疫系统,预防癌症的发生。但是,操纵个体细胞要比大群体难得多,因为每个细胞只产生少量的RNA,所以没有出错的余地。不仅仅是因为使用的工具,另一个问题是分析方法可能会导致大量数据的展示不够直观。由于数据文件从一个软件包传递到下一个软件包,每个工具都在处理一个步骤:基因组比对、质量控制和变异调用等,所以这个过程很复杂。但是对于批量RNA-seq来说,至少已经达成共识,哪些算法对每一步是最好的,以及它们应该如何运行。因此,现在存在的“流水线”即使不是完全即插即用,至少对于非专家来说也是非常容易处理的。但对于scRNA-seq而言,研究人员仍在研究他们可以对数据集做些什么,哪些算法是最有用的。但一系列在线资源和工具正在减轻scRNA-seq数据分析的工作量。 GitHub上一个名为“Awesome Single Cell”(go.nature.com/2rmb1hp)的页面包含了70多个工具和资源,涵盖了分析过程的每一步。
单细胞RNA-Seq(scRNA-seq)是指一类分析单个细胞转录组的方法。其中一些可能通过聚焦于3'或5'末端进行mRNA种类的筛选,而另一些则通过收集近全长序列来评估mRNA结构和剪接。单细胞分离策略涵盖人工细胞分选(起初用于微阵列研究)、基于流式细胞仪的分选、微流体装置,以及最近的基于微滴的和基于微孔的方法。当前连接到3'端计数的液滴和微孔进样具有最大的通量,允许在单个样品中同时快速处理数万个细胞。人们通常将scRNA-seq应用于新鲜解离的组织,但新兴的方案使用固定的细胞或从冷冻或轻微固定的组织分离的细胞核。固定或冷冻样品的应用将简化scRNA-seq的工艺流程,并为全面了解表达谱提供了可能。功效分析为比较这些方法的灵敏度和准确性提供了一个适用框架。
质量流式细胞术(CyTOF)和相关的方法允许基于用重金属条形码化的抗体进行蛋白质的多重测量。这些方法提供了预设的标签,并且需要针对每个目标的合适抗体,但是它们可以处理数百万个细胞,而且每个细胞所需的成本非常低。这些方法被应用于固定的细胞。最近,该方法已经扩展到通过用重金属标记的核酸探针的多重杂交来测量RNA特征了。
单细胞基因组和表观基因组测序可鉴定细胞基因组。基因组方法的目的是鉴定整个基因组或捕获特定的预定义区域。表观遗传学方法可以基于独特的组蛋白修饰(单细胞ChIP-Seq)、开放性(单细胞ATAC-Seq)、或同样鉴定DNA甲基化模式(单细胞DNAme-Seq)或3D组织(单细胞Hi -C)来捕获特定的预定义序列。目前人们已经使用组合条形码策略来捕获数万个单细胞。单细胞表观基因组学方法通常只研究细胞核,因此可以使用冷冻或某些固定的样品。由于基因组的大小和测序深度的限制,目前一些方法(如单细胞DNA测序)只能应用于相对较少的细胞。其它方法,如染色质组织的单细胞分析(通过单细胞ATAC-Seq)或单细胞ChIP-Seq产生的数据相对稀少,这说明对大量异形单元的分析既有挑战又有益处。计算分析已经可以通过将信号汇集到细胞之间以及跨基因组区域或基因座来解决这些问题。
单细胞组学的应用
单细胞组学可以有效判断某类细胞的分化和谱系追踪。细胞通过分化异步分支途径到达它们最终分化成的细胞类型。这是由分子变化所驱动,并反映在分子变化中的,特别是通过基因表达模式来完成的。因此,单细胞组学技术能将细胞演化过程发展重建为高维空间的轨迹,反映出Waddington的模式。只要观察到足够的细胞,人们甚至可以推断出一些突发事件。所需的采样密度将取决于路径和交叉点的数量和复杂性,良好的分类策略有助于分析稀有的细胞亚群。值得注意的是,沿发育路径不同点观察到的细胞的相对比例可以帮助传达关键信息,包括每个阶段的持续时间和祖细胞。
尽管我们仍然需要更好的算法,但是最初的计算方法已经能够用于从大量的单细胞分布图推断动态轨迹。关键的挑战包括准确推断分支结构,其中两个或更多个路径与单个点分离;重建“快速”转变,只有少数细胞可以被捕获;并且考虑到细胞可能同时遵循多个动态路径的事实。
线性发育轨迹已经能被重建,例如,从B细胞分化过程中的单细胞蛋白表达和体外成肌过程中的单细胞RNA表达、早期造血、体内神经发生和从成纤维细胞重编程为神经元等。分化的轨迹在胚胎干细胞、T辅助细胞和造血细胞的分化中也被重建,并且帮助人们解决了关于骨髓中的骨髓祖细胞是否已经倾向于不同命运这一悬而未决的问题。人类细胞图谱将成为研究疾病的重要参考,其涉及正常的细胞功能、相互作用、比例或生态系统的破坏。在数十年的组织病理学研究和FACS分析中,疾病的单细胞表现出的分析能力是显而易见的。它还将支持人们对理解异常细胞与组织生态系统中促进或抑制疾病过程(例如恶性细胞和肿瘤微环境之间)的所有其它细胞之间的相互作用。血液系统将是一个早期和富有成效的目标。基于单细胞组学技术对造血干细胞进行分析,我们可以更精确地分类急性髓细胞性白血病中的造血干细胞和造血祖细胞。此外,监测在正常情况下首次发现的罕见免疫人群也可以帮助消除疾病中的相关畸变。
短期内,受单细胞组学影响最大的可能是肿瘤。早期的研究使用单细胞qPCR来研究癌症干细胞的放射抗性的起源,并解析了结肠癌中细胞层次的异质性。随着高通量方法的出现,单细胞基因组分析已被用于研究乳腺癌和急性淋巴细胞白血病中肿瘤的克隆结构和进化,并推断引起急性骨髓性白血病的最早突变的顺序。
在恶性细胞中,单细胞组学技术可以鉴定出不同的细胞状态,如癌症干细胞、耐药状态、增殖和静止细胞,并且可以在个体患者中轻易地鉴定出单独的亚克隆。在非恶性细胞中,T细胞具有不同的功能状态,并且,尽管激活和耗竭的过程是耦合的,但是耗尽状态也由人类肿瘤中的独立调节程序控制。
参考文献
Single-cell biology. Nature. 547, 19 (06 July 2017) doi:10.1038/547019a
A Fantastic Voyage in Genomics. Science. 06 Oct 2017:Vol. 358, Issue 6359, pp. 56-57. DOI: 10.1126/science.358.6359.56
How to build a human cell atlas. Nature. 547, 24–26 (06 July 2017) doi:10.1038/547024a
Science Forum: The Human Cell Atlas. eLife 2017;6:e27041 DOI: 10.7554/eLife.27041
Single-cell sequencing made simple. Nature 547, 125–126 (06 July 2017) doi:10.1038/547125a



{kind=link}