
新工具助力单细胞表达数据的比较和分析
最近,科学家提出了两种可用于比较单细胞表达数据的方法,这些方法可以整合来自不同实验、不同条件的单细胞数据。
新的单细胞分子分析技术正在迅速改变生物医学研究。但单细胞数据分析存在一定的障碍,即不同平台的数据、同一平台的不同时间的数据、同一个样品不同实验方案的数据,以及同一个样品不同时间的数据间都存在的批次效应(batch effect),导致比较各个实验结果的数据的工作难度很大。近期Hemberg和Shen-Orr实验室分别提出了不同的方法,用于比较涉及不同条件、技术,甚至物种的实验的细胞样品。
单细胞RNA测序技术(single cell RNA sequencing, scRNA-seq)可以通过大量的单细胞生物信息学分析而获得生物学见解。许多研究都依靠维度降低技术来将数据2D或3D化。尽管这些方法揭示了细胞之间的相似性和差异性,但难以得到可量化的比较结果。此外,无监督聚类法(unsupervised clustering)通常基于基因表达谱的相似性而对单个细胞进行分类,并且帮助科学家解读群体异质性,例如确定新的细胞类型。但单个样本通常包含可能处于一个定向过程的不同阶段的异质细胞群体,例如正在分化或对扰动产生响应的细胞群体。目前科学家已经使用scRNA-seq技术来研究在这种动态过程中基因表达模式的变化,通过在虚拟时间轴(pseudotime axis)上对细胞进行计算排序,以重建该过程。
单细胞表达谱最令人兴奋的应用之一是比较不同状态下细胞基因表达的差异。这对于了解疾病和确定潜在的治疗靶标具有非常重要的意义。最近,研究人员开始将自己的数据与参考样本的数据进行比较,这也是最近多项计划,如人类细胞图谱计划(Human Cell Atlas initiative),正在努力推出数据集的标准参考数据的原因。虽然将来自多个实验的scRNA-seq数据结合起来意义非凡,但由于样品来源、制备和测序的差异,而非细胞状态造成的差异,使得整合来自不同试验的单细胞测序数据变得非常困难。
Kiselev等人提出了一种将细胞实验数据与注释参考数据相互联系起来的方法(图1a)。他们的算法名为scmap-cluster,它分析了待测样本的基因表达的空间距离,以便将细胞与参考数据中最相似的细胞匹配起来。scmap首先会选定执行计算所需的一系列特征指标。有趣的是,研究人员发现,相比于选择高度可变或随机的基因作为参考,选择期望表达频率高于0的基因能产生更准确的映射,这一发现对于其它类型的scRNA-seq数据分析也可能是有意义的。尽管该算法尝试将细胞与参考集相匹配,但如果细胞的基因表达模式与参考数据差异较大,那么细胞便不会被归类。这是一个值得认真考虑的因素,因为不同实验中的细胞类型可能不完全相同。Kiselev等人向用户提供了R安装包(R package)和Web版本,并确保算法能在大型数据集上快速运行,这种做法非常值得学习。
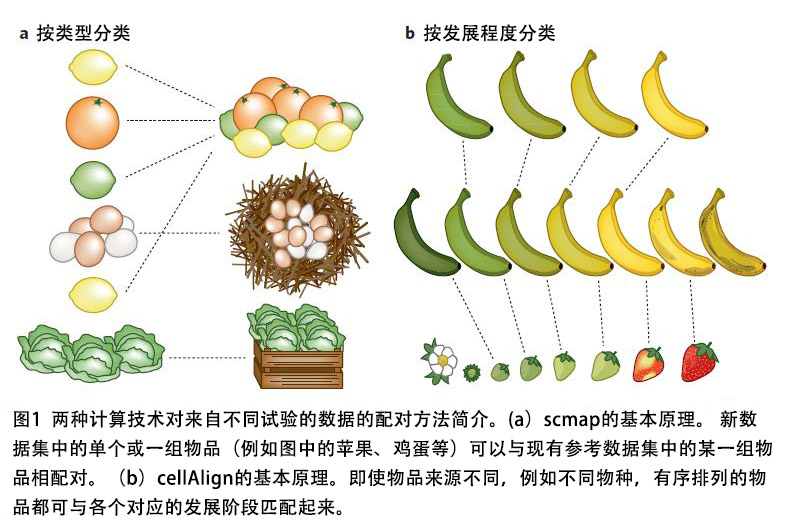
由于离散聚类不容易捕捉到分化过程的连续过程,Kiselev等人又提出了一种近邻取样方法,即将细胞与非聚类(例如虚拟时间轴排序)的参考数据集匹配起来。
为了更深入地比较虚拟时间轴排序,Alpert等人开发了cellAlign算法。cellAlign使用动态时间来将待测样本的动态表达谱与参考数据集的动态表达谱匹配起来,从而实现动态表达模式的比较(图1b)。令人兴奋的是,cellAlign不仅能够比较整个转录组,还可以利用特定的基因或基因模块来评估实验条件之间的差异。Alpert等人甚至分析了来自植入前胚胎的scRNA-seq数据,以鉴定人类胚胎和小鼠胚胎在发育过程中的基因模块表达模式之间的差异,由此证明其算法在比较来自不同来源的数据上的能力。
由于scmap和cellAlign的目的不同,前者更注重聚类,而后者更注重动态变化,因此选择哪一种算法将取决于所研究的问题。值得注意的是,这两种方法都不以“批量修正”数据为目的,而是以帮助下游分析,如降低综合数据集的维度为目的。Satija和Marioni的实验室也提出了一种类似的算法,其适用于特定目的,例如寻找在不同条件下差异表达的基因。此外,科学家也在研究虚拟空间排序算法。该算法不是按时间进度排序,而是按照单个细胞的基因表达来推算其位置坐标,然后进行位置排序。
不管是scmap,还是cellAlign算法,它们的优点都在于揭示扰动是如何影响基因转录谱的,特别是在疾病的情况下。比较分析来自人类或来自小鼠模型的受干扰细胞群与其野生型对照组,应该能够发现哪些细胞群体或分化阶段受到最大的影响,以及基因表达发生了哪些改变。
比较通过不同方案产生的数据时,其中一个困难是解决不同方法所固有的技术特性的差异,例如每个细胞中检测到的基因数目的巨大差异。Kiselev等人和Alpert等人都简单地介绍了这一点:scmap的创建者指出,他们的算法通过努力寻找离零表达基因(通常是由于数据库生成期间遗失或捕获失败)最近的细胞来克服这一障碍,cellAlign的开发者则讨论了由于数据中的技术差异而需要扩展基因表达的问题。未来还需要进一步探索如何在scRNA-seq领域可靠地比较不同技术产生的不同数据集。大规模整合数据集的计划,如人类细胞图谱计划,肯定会有助于推动该领域的进一步创新。
原文检索:
Liam Drew. (2018) Fighting the inevitability of ageing. Nature, 555: S15-S17.
张洁/编译



{kind=link}