
基因筛查进入单细胞时代
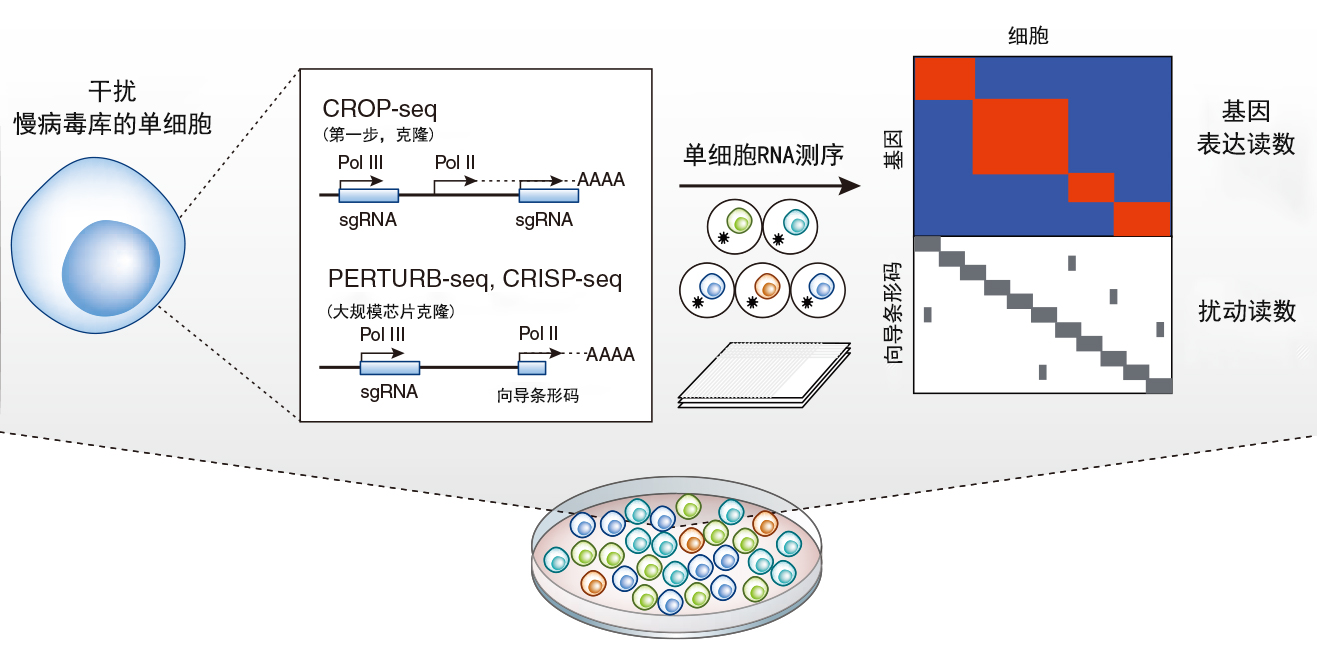
图1 使用混合CRISPR扰动和scRNA-seq的大规模并行基因筛查。两种方法使用两个小向导RNA表达策略向单个细胞导入扰动载体。scRNA-seq可以同时检测扰动和详细的转录组表型。
四项研究通过整合单细胞转录组学,克服了基因筛查的局限性。
遗传筛选是生物学中最有力的工具,它可以阐明复杂的生物过程。最近四项研究已经开发了一个新的遗传分析方法,通过使用单个细胞作为微观实验室,测定扰动。这些研究开发了CROP-seq、PERTURB-seq和CRISP-seq技术,克服了现有基因筛查方法的局限性,并且可以分析一些原本无法分析的样本。
目前,若要将遗传扰动与细胞表型相关联,人们主要使用两种策略——芯片筛查和混合筛查,但两种方法都各有缺点。芯片筛查中,细胞接种于微孔板中,每个微孔中引入单个扰动,并基于成像、转录或蛋白质的测量,提供每个孔中细胞的高分辨率表型。然而,芯片筛查受到高成本和劳动力和/或机器人的可用性的限制。并且每次筛查,需要的细胞数量较多,因此通常只能分析可在培养皿中增殖的细胞。相比之下,混合筛查允许在单个细胞样本中快速筛查数千个并行扰动。在混合筛查中,扰动通过与单个特定表型(例如细胞存活)相关联来排序。不同于芯片筛查,混合筛查不会显示每个扰动的详细表型;并且可靠的筛查标志物还有待开发。由于假阳性率高,混合筛查结果通常还需要二次筛查来验证。
CROP-seq、CRISP-seq和Perturb-seq技术结合了两种筛查策略的优势,代表的是功能基因组学的根本转变。类似于混合筛查,这些新方法使用CRISPR-Cas9系统在单个样本中并行生成多达数千个遗传扰动,但使用单细胞RNA-seq(single cell RNA-seq, scRNA-seq)作为读数。该方法可以同时测量每个细胞的扰动和表型(图1)。这些新技术不依赖于特定表型或存活,表型被记录为整个转录组,为解剖基因功能关系提供了惊人的数据。基于板的scRNA-seq平台还可以为每个细胞进行附加测量,如成像或FACS数据。所有这些新方法都成功地将丰富的表型信息与细胞平板中的每个特定扰动相关联,有助于实现大规模基因组功能筛查。为了解读这些技术得到的数据,每个研究小组开发了线性回归模型,由Dixit等人进一步形式化。
将CRISPR筛选融入scRNA-seq,在技术上并不容易。CRISPR驱动的扰动需要从RNA聚合酶III(Pol III)特异性U6启动子处,表达高水平的靶向特定基因的单向导RNA(single guide RNA, sgRNA)。但是sgRNA缺少poly(A)尾端,所以其身份不能通过标准scRNA-seq方法读出。所有这三种方法通过产生携带pol III:sgRNA和Pol II驱动的可选择和/或荧光标记的载体来解决这个读出问题,其中3'UTR含有sgRNA特异性序列。PERTURB-seq和CRISP-seq依赖于芯片克隆策略产生的RNA库,来将每个sgRNA链接到特定的扰动条形码转录物上。相比之下,CROP-seq的克隆解决方案更为简单优雅:将Pol III:sgRNA复合体整合到报告转录物中,并插入到慢病毒载体的长重复序列末端中。这样Pol III:sgRNA复合体在病毒整合期间会被复制。这种简化的克隆方法使得CROP-seq与现有的CRISPR筛查sgRNA库融合在一起。
这些论文提出的方法无需生物标志物,可用于检测之前无法进行的检测(如混合检测、复杂细胞群体中的扰动检测,以及无法培育增殖的稀有细胞的筛查)。所有研究表明,这些方法可以很容易地识别单个细胞中的多个扰动。Datlinger等人、Dixit等人和Adamson等人的研究证明了这些方法可用于检测单一细胞类型或同系培养细胞的扰动。Jaitin等人证明,扰动也可以在更复杂的场景中进行,包括活的小鼠体内。该实验特别令人印象深刻,他们从GFP-Cas9小鼠中取得造血祖细胞,用sgRNA载体的混合物转入向导RNA,并注射到次级受体小鼠中。在成功植入后,这些小鼠被免疫攻击,研究者取出脾细胞,并且使用CRISP-seq检测每个扰动的影响——受刺激的细胞偏向于分化成某些骨髓细胞的程度或改变激活程序,对刺激物产生响应。
这些平台的一个特别的优点是完整的转录组测序提供了“一刀切”的测定法,可同时捕获多种表型,而不依赖生物标志物。这些测定还利用CRISPR-Cas9系统的模块化和灵活性。CRISPR-Cas9系统不仅可以用于基因敲除,还能用于基因沉默和基因激活。Adamson等人使用靶向多达18,905个基因的向导RNA库,证明了CRISPRi的可靠性和可扩展性。Dixit等人分析了超过20万个细胞,证明了单细胞转录组学的可扩展性。相反,将来的技术可能直接对一小份活检样本中的几万个细胞进行混合筛查。
当然,这些方法也不乏局限性。也许最根本的是,信息表型很大程度上限于细胞自主过程,而许多过程,如干细胞分化,则依赖细胞信号传导。剖析非自主过程仍然需要使用大规模筛查。另一个限问题是需要确定每个细胞中每个基因是否被成功敲除。因为技术报告指出sgRNA存在,但不能直接显示这些sgRNA是否确实发挥了作用。然而,如Adamson等人所证明的,通过分析转录读数可以部分解决这个问题。另一个实际的问题是成本。虽然增加每个细胞的测序深度通常可以克服scRNA-seq数据的固有噪声,但是为了节约成本,就只能减少检测的细胞数目。Datlinger等人和Jaitin等人结论分别是,大约30个细胞和50-100个细胞就足以捕获每个扰动表型。尽管有这些局限性,但是CROP-seq、CRISP-seq和PERTURB-seq的简单性以及它们带来的新应用在未来几年将大有可为。
原文检索:
Daniel E Wagner & Allon M Klein. (2017) Genetic screening enters the single-cell era. Nature Methods, 14(1038): 237-238.
张洁/编译



{kind=link}